Abbott, R.J., Smith, L.C., Milne, R.I., Crawford, R.M., Wolff, K., Balfour, J., 2000. Molecular analysis of plant migration and refugia in the Arctic. Science 289, 1343-1346.
|
Backström, N., Brandström, M., Gustafsson, L., Qvarnström, A., Cheng, H., Ellegren, H., 2006. Genetic mapping in a natural population of collared flycatchers ( Ficedula albicollis): conserved synteny but gene order rearrangements on the avian Z chromosome. Genetics 174, 377-386.
|
Benjamini, Y., Hochberg, Y., 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. -B (Method. ) 57, 289-300.
|
Bertorelle, G., Raffini, F., Bosse, M., Bortoluzzi, C., Iannucci, A., Trucchi, E., et al., 2022. Genetic load: genomic estimates and applications in non-model animals. Nat. Rev. Genet. 23, 492-503.
|
Bolger, A.M., Lohse, M., Usadel, B., 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114-2120.
|
Ceballos, F.C., Joshi, P.K., Clark, D.W., Ramsay, M., Wilson, J.F., 2018. Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 19, 220.
|
Charlesworth, D., Willis, J.H., 2009. The genetics of inbreeding depression. Nat. Rev. Genet. 10, 783-796.
|
Danecek, P., Auton, A., Abecasis, G., Albers, C.A., Banks, E., DePristo, M.A., et al., 2011. The variant call format and VCFtools. Bioinformatics 27, 2156-2158.
|
Dussex, N., Van Der Valk, T., Morales, H.E., Wheat, C.W., Díez-del-Molino, D., Von Seth, J., et al., 2021. Population genomics of the critically endangered kākāpō. Cell Genom. 1, 100002.
|
Fay, J.C., Wyckoff, G.J., Wu, C.-I., 2001. Positive and negative selection on the human genome. Genetics 158, 1227-1234.
|
Feng, S., Fang, Q., Barnett, R., Li, C., Han, S., Kuhlwilm, M., et al., 2019. The genomic footprints of the fall and recovery of the crested ibis. Curr. Biol. 29, 340-349. e347.
|
Frankham, R., 1995a. Conservation genetics. Ann. Rev. Genet. 29, 305-327.
|
Frankham, R., 1995b. Inbreeding and extinction: a threshold effect. Conserv. Biol. 9, 792-799.
|
García-Dorado, A., 2012. Understanding and predicting the fitness decline of shrunk populations: inbreeding, purging, mutation, and standard selection. Genetics 190, 1461-1476.
|
Grossen, C., Guillaume, F., Keller, L.F., Croll, D., 2020. Purging of highly deleterious mutations through severe bottlenecks in Alpine ibex. Nat. Comm. 11, 1001.
|
Hale, M., Jensen, H., Birkhead, T., Burke, T., Slate, J., 2008. A comparison of synteny and gene order on the homologue of chicken chromosome 7 between two passerine species and between passerines and chicken. Cytogenet. Genom. Res. 121, 120-129.
|
Hedrick, P., Robinson, J., Peterson, R.O., Vucetich, J.A., 2019. Genetics and extinction and the example of Isle Royale wolves. Anim. Conserv. 22, 302-309.
|
Hedrick, P.W., 1994. Purging inbreeding depression and the probability of extinction: full-sib mating. Heredity 73, 363-372.
|
Hedrick, P.W., Garcia-Dorado, A., 2016. Understanding inbreeding depression, purging, and genetic rescue. Trends Ecol. Evol. 31, 940-952.
|
|
Höglund, J., 2009. Evolutionary Conservation Genetics. Oxford University Press, Oxford.
|
|
Ingolfsson, O., Moller, P., Lubinski, D., Forman, S., 2006. Severnaya Zemlya, Arctic Russia: a Nucleation Area for Kara Sea Ice Sheets During the Middle to Late Quaternary. AGU Fall Meeting Abstracts.
|
IUCN, 2020. Version 2019-3. The IUCN red list of threatened species. .
|
|
Kardos, M., Åkesson, M., Fountain, T., Flagstad, Ø., Liberg, O., Olason, P., et al., 2018. Genomic consequences of intensive inbreeding in an isolated wolf population. Nat. Ecol. Evol. 2, 124-131.
|
Kardos, M., Armstrong, E.E., Fitzpatrick, S.W., Hauser, S., Hedrick, P.W., Miller, J.M., et al., 2021. The crucial role of genome-wide genetic variation in conservation. Proc. Natl. Acad. Sci. USA 118, e2104642118.
|
Keller, L.F., Waller, D.M., 2002. Inbreeding effects in wild populations. Trends Ecol. Evol. 17, 230-241.
|
Khan, A., Patel, K., Shukla, H., Viswanathan, A., van der Valk, T., Borthakur, U., et al., 2021. Genomic evidence for inbreeding depression and purging of deleterious genetic variation in Indian tigers. Proc. Natl. Acad. Sci. USA 118, e2023018118.
|
Kyriazis, C.C., Wayne, R.K., Lohmueller, K.E., 2021. Strongly deleterious mutations are a primary determinant of extinction risk due to inbreeding depression. Evol. Lett. 5, 33-47.
|
Li, H., 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987-2993.
|
|
Li, H., 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997.
|
Li, H., Durbin, R., 2011. Inference of human population history from individual whole-genome sequences. Nature 475, 493.
|
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al., 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078-2079.
|
Lynch, M., Gabriel, W., 1990. Mutation load and the survival of small populations. Evolution 44, 1725-1737.
|
|
Mathur, S., Tomeček, J.M., Tarango-Arámbula, L.A., Perez, R.M., DeWoody, J.A., 2022. An evolutionary perspective on genetic load in small, isolated populations as informed by whole genome resequencing and forward-time simulations. Evolution 77, 690-704.
|
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al., 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genom. Res. 20, 1297-1303.
|
McLaren, W., Gil, L., Hunt, S.E., Riat, H.S., Ritchie, G.R., Thormann, A., et al., 2016. The ensembl variant effect predictor. Genom. Biol. 17, 122.
|
Mooney, J.A., Marsden, C.D., Yohannes, A., Wayne, R.K., Lohmueller, K.E., 2023. Long-term small population size, deleterious variation, and altitude adaptation in the Ethiopian wolf, a severely endangered canid. Mol. Biol. Evol. 40, msac277.
|
Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., Durbin, R., 2016. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749-1751.
|
Newman, D., Pilson, D., 1997. Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evolution 51, 354-362.
|
Ng, P.C., Henikoff, S., 2003. SIFT: Predicting amino acid changes that affect protein function. Nucl. Acids Res. 31, 3812-3814.
|
Persons, N.W., Hosner, P.A., Meiklejohn, K.A., Braun, E.L., Kimball, R.T., 2016. Sorting out relationships among the grouse and ptarmigan using intron, mitochondrial, and ultra-conserved element sequences. Mol. Phylogenet. Evol. 98: 123-132.
|
Robinson, J.A., Brown, C., Kim, B.Y., Lohmueller, K.E., Wayne, R.K., 2018. Purging of strongly deleterious mutations explains long-term persistence and absence of inbreeding depression in island foxes. Curr. Biol. 28, 3487-3494.
|
Rózsa, J., Strand, T.M., Montadert, M., Kozma, R., Höglund, J., 2016. Effects of a range expansion on adaptive and neutral genetic diversity in dispersal limited Hazel grouse (Bonasa bonasia) in the French Alps. Conserv. Genet. 17, 401-412.
|
|
Saarnisto, M., Lunkka, J.P., 2004. Climate variability during the last interglacial-glacial cycle in NW Eurasia. In: Battarbee, R.W., Gasse, F., Stickley, C.E. (Eds.), Past Climate Variability through Europe and Africa, vol. 6. Springer, Dordrecht, pp. 443–464.
|
Schiffels, S., Durbin, R., 2014. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46, 919.
|
|
Shields, W.M., 1987. Dispersal and mating systems: investigating their causal connections. In: Chepko-Sad, B.D., Halpin, Z.T. (Eds.), Mammalian Dispersal Patterns: the Effects of Social Structure on Population Genetics. University of Chicago Press, Chicago, pp. 3–24.
|
|
Smeds, L., Ellegren, H., 2022. From high masked to high realized genetic load in inbred Scandinavian wolves. Mol. Ecol. 32, 1567-1580.
|
Song, K., Gao, B., Halvarsson, P., Fang, Y., Jiang, Y.-X., Sun, Y.-H., et al., 2020. Genomic analysis of demographic history and ecological niche modeling in the endangered Chinese Grouse Tetrastes sewerzowi. BMC Genom. 21, 581.
|
Song, K., Gao, B., Halvarsson, P., Fang, Y., Klaus, S., Jiang, Y.-X., et al., 2021. Demographic history and divergence of sibling grouse species inferred from whole genome sequencing reveal past effects of climate change. BMC Ecol. Evol. 21, 1-10.
|
|
Soulé, M.E., 1980. Conservation Biology: An Evolutionary Ecological Perspective. Sinauer Associates Inc., Sunderland, MA.
|
Storch, I., 2000. Conservation status and threats to grouse worldwide: an overview. Wildl. Biol. 6, 195-205.
|
Sun, Y.-H., 2000. Distribution and status of the Chinese grouse Bonasa sewerzowi. Wildl. Biol. 6, 271-275.
|
Sun, Y.H., Swenson, J.E., Fang, Y., Klaus, S., Scherzinger, W., 2003. Population ecology of the Chinese grouse, Bonasa sewerzowi, in a fragmented landscape. Biol. Conserv. 110, 177-184.
|
Swenson, J.E., 1991. Status and conservation of hazel grouse in Europe. Ornis Scand. 22, 297-298.
|
Szpiech, Z.A., Xu, J., Pemberton, T.J., Peng, W., Zöllner, S., Rosenberg, N.A., et al., 2013. Long runs of homozygosity are enriched for deleterious variation. Am. J. Human Genet. 93, 90-102.
|
Tallmon, D.A., Luikart, G., Waples, R.S., 2004. The alluring simplicity and complex reality of genetic rescue. Trends Ecol. Evol. 19, 489-496.
|
|
Van der Auwera, G.A., Carneiro, M.O., Hartl, C., Poplin, R., Del Angel, G., LevyMoonshine, A., et al., 2013. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43 (11), 10.11-11.10.33.
|
|
van der Valk, T., 2019. Genomics of Population Decline. Doctoral Dissertation. Uppsala Unversity, Uppsala.
|
van der Valk, T., de Manuel, M., Marques-Bonet, T., Guschanski, K., 2019a. Estimates of genetic load in small populations suggest extensive purging of deleterious alleles. bioRxiv. .
|
van der Valk, T., Díez-del-Molino, D., Marques-Bonet, T., Guschanski, K., Dalén, L., 2019b. Historical genomes reveal the genomic consequences of recent population decline in eastern gorillas. Curr. Biol. 29, 165-170.
|
van Oosterhout, C., 2020. Mutation load is the spectre of species conservation. Nat. Ecol. Evol. 4, 1004-1006.
|
|
Vaurie, C., 1965. The birds of the Palearctic fauna. Non-passeriformes. Bird-Banding 37, 145-146.
|
Warren, W.C., Hillier, L.W., Tomlinson, C., Minx, P., Kremitzki, M., Graves, T., et al., 2017. A new chicken genome assembly provides insight into avian genome structure. G3 Genes Genom. Genet. 7, 109-117.
|
Wootton, E., Robert, C., Taillon, J., Côté, S., Shafer, A.B., 2023. Genomic health is dependent on long-term population demographic history. Mol. Ecol. 32, 1943-1954.
|
|
Wright, S., 1977. Evolution and the Genetics of Populations. 2. The Theory of Gene Frequencies. University of Chicago Press, Chicago, IL.
|
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al., 2011. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucl. Acids Res. 39 (suppl_2), W316–W322.
|
Xue, Y., Prado-Martinez, J., Sudmant, P.H., Narasimhan, V., Ayub, Q., Szpak, M., et al., 2015. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348, 242-245.
|
Zhan, X., Zheng, Y., Wei, F., Bruford, M.W., Jia, C., 2011. Molecular evidence for Pleistocene refugia at the eastern edge of the Tibetan Plateau. Mol. Ecol. 20, 3014-3026.
|

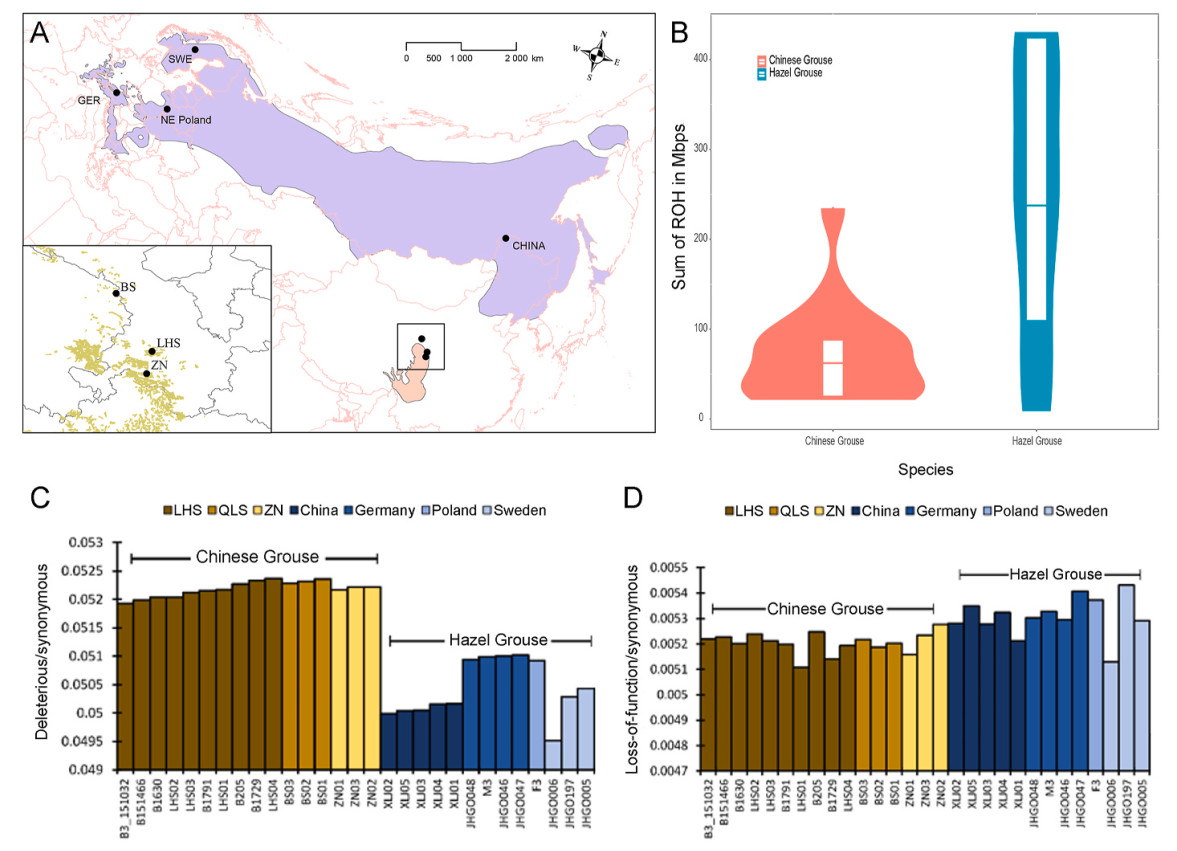
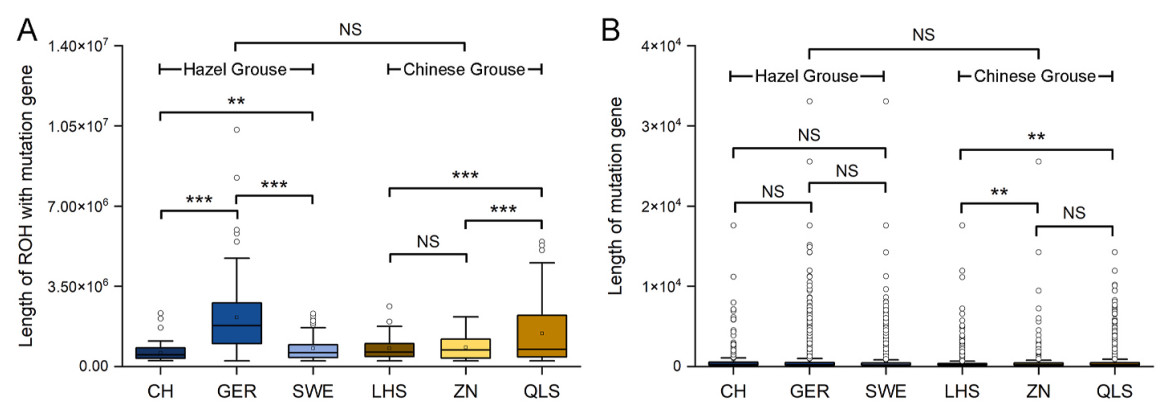
 DownLoad:
DownLoad:





 Email Alerts
Email Alerts RSS Feeds
RSS Feeds