Per Alström, Zeinolabedin Mohammadi, Erik D. Enbody, Martin Irestedt, Derek Engelbrecht, Pierre-André Crochet, Alban Guillaumet, Loïs Rancilhac, B. Irene Tieleman, Urban Olsson, Paul F. Donald, Martin Stervander. 2023: Systematics of the avian family Alaudidae using multilocus and genomic data. Avian Research, 14(1): 100095. DOI: 10.1016/j.avrs.2023.100095
Citation:
Per Alström, Zeinolabedin Mohammadi, Erik D. Enbody, Martin Irestedt, Derek Engelbrecht, Pierre-André Crochet, Alban Guillaumet, Loïs Rancilhac, B. Irene Tieleman, Urban Olsson, Paul F. Donald, Martin Stervander. 2023: Systematics of the avian family Alaudidae using multilocus and genomic data. Avian Research, 14(1): 100095. DOI: 10.1016/j.avrs.2023.100095
Per Alström, Zeinolabedin Mohammadi, Erik D. Enbody, Martin Irestedt, Derek Engelbrecht, Pierre-André Crochet, Alban Guillaumet, Loïs Rancilhac, B. Irene Tieleman, Urban Olsson, Paul F. Donald, Martin Stervander. 2023: Systematics of the avian family Alaudidae using multilocus and genomic data. Avian Research, 14(1): 100095. DOI: 10.1016/j.avrs.2023.100095
Citation:
Per Alström, Zeinolabedin Mohammadi, Erik D. Enbody, Martin Irestedt, Derek Engelbrecht, Pierre-André Crochet, Alban Guillaumet, Loïs Rancilhac, B. Irene Tieleman, Urban Olsson, Paul F. Donald, Martin Stervander. 2023: Systematics of the avian family Alaudidae using multilocus and genomic data. Avian Research, 14(1): 100095. DOI: 10.1016/j.avrs.2023.100095
Animal Ecology, Department of Ecology and Genetics, Evolutionary Biology Centre, Uppsala University, Norbyvägen 18D, SE-752 36,
Uppsala, Sweden. E-mail address: per.alstrom@ebc.uu.se (P. Alström)
Bird Group, Natural History Museum, Akeman Street, Tring, Hertfordshire, HP23 6AP, United Kingdom. E-mail address: martin@stervander.com (M. Stervander)
The family Alaudidae, larks, comprises 93–100 species (depending on taxonomy) that are widely distributed across Africa and Eurasia, with single species extending their ranges to North and northernmost South America and Australia. A decade-old molecular phylogeny, comprising ~80% of the species, revealed multiple cases of parallel evolution and large variation in rates of morphological evolution, which had misled taxonomists into creating many non-monophyletic genera. Here, we reconstruct the phylogeny of the larks, using a dataset covering one mitochondrial and 16 nuclear loci and comprising all except one of the currently recognised species as well as several recently proposed new species (in total 133 taxa; not all loci available for all species). We provide additional support using genome-wide markers to infer a genus-level phylogeny based on near-complete generic sampling (in total 51 samples of 44 taxa across 40 species). Our results confirm the previous findings of rampant morphological convergence and divergence, and reveal new cases of paraphyletic genera. We propose a new subfamily classification, and also that the genus Mirafra is divided into four genera to produce a more balanced generic classification of the Alaudidae. Our study supports recently proposed species splits as well as some recent lumps, while also questioning some of the latter. This comprehensive phylogeny will form an important basis for future studies, such as comparative studies of lark natural history, ecology, evolution and conservation.
The family Alaudidae, larks, is part of the superfamily Sylvioidea, which also includes various groups given the common names warblers, babblers, hirundines, bulbuls etc., as well as the single species that is sister to the larks, the Bearded Reedling (Panurus biarmicus) (Alström et al., 2006, 2013b; Fregin et al., 2012; Oliveros et al., 2019). The number of currently recognised species of larks varies from 93 (Clements et al., 2022) to 100 (Gill et al., 2022), although both these authorities recognise the same 21 genera. The discrepancy between these two main checklists concern species recognised by Gill et al. (2022) that have recently (October 2022) been lumped by Clements et al. (2022). The larks are widely distributed across Africa and Eurasia, with 59–60% of the species and 33% of the genera endemic to Africa. A single lineage (the Horned Lark Eremophila alpestris complex) has reached North America and Colombia from its ancestral Eurasian range (Ghorbani et al., 2020a), and another species (Horsfield’s Bushlark Mirafa javanica) has reached Australia from Africa and southern Asia (Alström et al., 2013a). Larks inhabit a broad range of open habitats, including short-grass habitats, agricultural lands, open scrublands, stony as well as sandy deserts and tundra (de Juana et al., 2004; Fjeldså et al., 2020a). A few species occur in glades and clearings in open forest and along forest edges (de Juana et al., 2004). Most species are cryptically coloured and sexually monomorphic, and superficially similar in appearance. Males of Black Lark (Melanocorypha yeltoniensis) and all except one of the species of sparrow-larks Eremopterix species are the most notable exceptions to this rule (de Juana et al., 2004; Fjeldså et al., 2020a). Lark plumages often match the colour of the substrates they inhabit, which may be adaptive as it can increase camouflage (Donald et al., 2017; Mason et al., 2023). Many larks have elaborate songs that often include outstanding mimicry, but some species have very simple songs (de Juana et al., 2004). A molecular phylogeny for the Alaudidae revealed that morphological evolution was not tightly associated with genetic divergence (Alström et al., 2013a). Instead, the authors found multiple instances of remarkable convergent evolution in distant lineages, cases of deep genetic divergence between taxa that were treated as conspecific, as well as other instances of extraordinary morphological divergence between closely related species.
The number of recognised species of larks has increased remarkably over the past six decades (Peters, 1960, 76 species; Clements et al., 2022, 93 species; Gill et al., 2022, 100 species). This increase is mainly due to studies of, in particular, molecular data and/or songs that have resulted in the elevation of several taxa from the rank of subspecies to species (Alström, 1998; Ryan et al., 1998; Ryan and Bloomer, 1999; Guillaumet et al., 2005, 2006, 2008; Stervander et al., 2016, 2020a; Donald and Christodoulides, 2018; Ghorbani et al., 2020a; Alström and Sundev, 2021; Alström et al., 2021). Other splits have been proposed, but not yet endorsed by the main checklists (Drovetski et al., 2014; Ghorbani et al., 2020b), while a few lumps have also been proposed and accepted (Donald and Collar, 2011; Spottiswoode et al., 2013). The recent (25 October 2022) checklist by Clements et al. (2022), which is the one we follow as our baseline taxonomy in this paper, lumped eight of the species recognised by Gill et al. (2022) while it split one species (Table 1). It nevertheless seems likely that the number of lark species will increase as a result of studies that integrate different categories of data (e.g., morphological, vocal, genetic and behavioural data). This is particularly true for Africa, which has several lark species with multiple poorly known subspecies. For example, a recent integrative taxonomic study of the African Rufous-naped Lark (Mirafra africana)–Sharpe’s Lark (M. sharpii)–Red-winged Lark (M. hypermetra)–Somali Lark (M. somalica)–Ash’s Lark (M. ashi) species complex proposed recognition of nine instead of five species (Alström et al., submitted).
Table
1.
Proposed species-level taxonomy for the family Alaudidae, comprising all lark taxa included in the present study in linear sequence (following the principles of Fjeldså et al., 2020b), compared to the taxonomy according to Gill et al. (2022) and Clements et al. (2022). In cases where the latter two are conflicting, the treatment supported by our study is highlighted in green font and the unsupported one in red font. Changes in our proposed taxonomy are highlighted in blue font; tentative but uncertain species status in purple font. The family, as defined here, contains 103 species across three subfamilies.
Only one comprehensive molecular phylogeny, based on mitochondrial DNA (mtDNA) and three nuclear loci and comprising ~80% of the species, has previously been published for the Alaudidae (Alström et al., 2013a). It revealed an exceptionally high level of disagreement between the traditional, morphology-based, taxonomy and phylogenetic relationships, and resulted in several taxonomic changes. Here, we expand the sampling of mtDNA and nuclear loci to include all except one of the currently recognised species (99%), as well as all of the species proposed by Ghorbani et al. (2020b) and Alström et al., (submitted). We further assess the resulting phylogenetic hypothesis by constructing a genus-level phylogeny using genome-wide markers by sampling representatives for all but three genera. The resulting phylogeny forms the basis for a taxonomic revision and provides a valuable framework for future comparative studies of various aspects of lark evolution and conservation.
2.
Methods
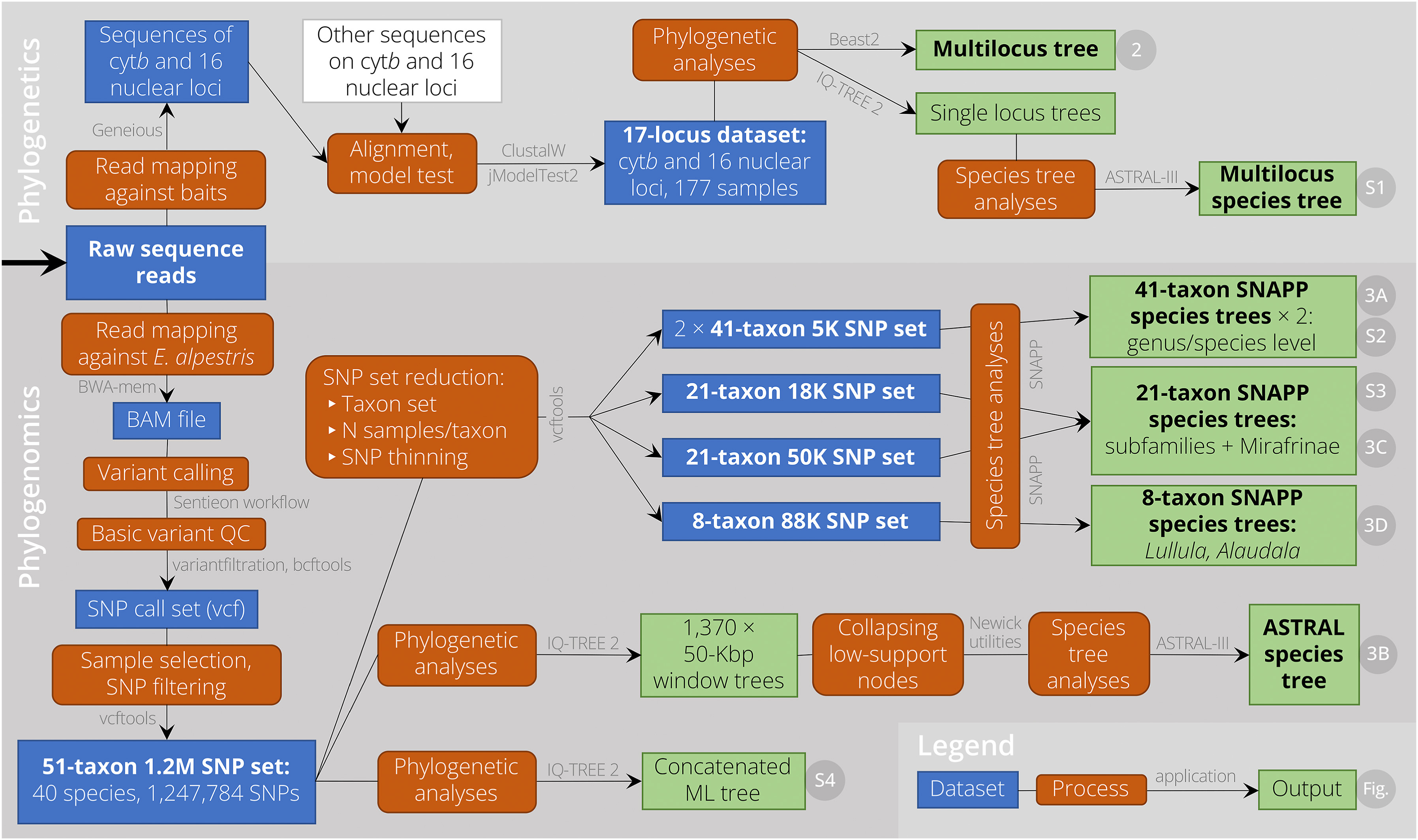
The datasets, analyses, and output are graphically summarised in Fig. 1, while details are provided below.
Figure
1.
Flow chart detailing the major datasets, processes, and applications used in this study. Further, it shows the main output, and in case the output is included as a figure, the figure number. A key is located in the bottom right corner; additionally, the white box represents external material, and bold font indicates main components referred to in the manuscript. Details on all analyses are available in the Methods section.
Our sampling scheme follows the taxonomy of Clements et al. (2022). We obtained fresh or toepad samples for 38 lark taxa, for which whole genomes were resequenced (see below and Appendix Table S1). Comparable data were obtained for another 21 samples of 17 Mirafra lark taxa from Alström et al., and for the outgroup Panurus biarmicus (Short Read Archive accession SRX7050291) from Sigeman et al. (2020). In addition, 306 sequences from 111 taxa from Alström et al. (2013a) and various other sources were downloaded from GenBank (Appendix Table S1). In all, our sampling comprises 184 samples of 133 lark taxa and includes all except one of the species currently recognised in Alaudidae (the poorly known Friedmann’s Lark Mirafra pulpa). The genetic sampling (Appendix Table S1) varied from one gene (mitochondrial cytochrome b, cytb; 19 species) to >1 million single nucleotide polymorphisms (SNPs) (39 species).
For the fresh samples, DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). For museum toepad samples, we followed existing protocols of laboratory procedures for degraded DNA, including all procedures from DNA extraction, USER enzyme (New England Biolabs) treatment, and library constructions in Irestedt et al. (2022) and Meyer and Kircher (2010). Whole genomes were resequenced at ~10–43.5x coverage (mean 22.1x ± 9.4 s.d.) on an Illumina NovaSeq 6000 S4 (Illumina, CA) at SciLifeLab, Stockholm and Uppsala.
The mitochondrial cytb, nuclear myoglobin intron 2 and partial exons (Myo), ornithine decarboxylase introns 6–7 and partial exons (ODC) and recombination activating gene part 1 (RAG1) were harvested from 57 resequenced genomes (an additional 10 for cytb only; Appendix Table S1). This was done by mapping short reads of each species to reference sequences of these loci, which were obtained from the Horned Lark (Eremophila alpestris) reference genome, using Geneious v.9.1.2 (Biomatters Ltd), with the “map to reference” command. This process was then repeated, with the consensus sequence of the assembled reads of each species used as a template for that same species. From 35 resequenced genomes (Appendix Table S1), 13 additional genetic markers were harvested the same way: aconitase 1 mainly intron 9 (ACO1), brahma protein, mainly intron 15 (BRM), chromodomain helicase DNA binding protein 1 (CHD1z), beta-fibrinogen mainly intron 5 (Fib5) and mainly intron 7 (Fib7), glyceraldehyde 3-phosphate mainly intron 11 (G3P), interphotoreceptor retinoid-binding protein (IRBP), L-lactate dehydrogenase mainly intron 3 (LDH), melanocortin 1 receptor complete codons (MC1R), rhodopsin mainly intron 1 (RHO), transforming growth factor beta 2 mainly intron 5 (TGFb2), early growth response protein exon 2 (ZENK) and 3′ untranslated region (ZENKUTR). All new sequences have been submitted to GenBank (Appendix Table S1).
We called variants from 50 resequenced lark samples and the outgroup Bearded Reedling (Panurus biarmicus) (Appendix Table S1), relative to the Eremophila alpestris reference genome (GCA_009792885.1; Mason et al., 2020). We first trimmed Illumina adapters using the software Fastp v. 0.20.0 (Chen et al., 2018) and evaluated sequence quality using FastQC (Andrews, 2010). Read mapping and variant calling were performed using the accelerated wrapper tools in the Sentieon toolkit v. 201911 (Freed et al., 2017). Reads were mapped to the Eremophila alpetris genome using BWA-mem v. 0.7.17-r1188 (Li, 2013) and PCR duplicates removed using the LocusCollector and dedup modules in Sentieon. We called variable positions using HaplotypeCaller GATK v. 4.1 (McKenna et al., 2010) and performed joint genotyping of all samples using GenotypeGVCFs (GATK v. 4.1) by using the Haplotyper and Genotyper modules, respectively, in Sentieon. Finally, we applied the following common GATK filters to SNPs using the VariantFiltration command in GATK v. 4.1: RPRS < −8, QD < 2.0, FS > 60.0, SOR > 3.0, MQ < 40.0, and MQRankSum < −12.5. We additionally set genotypes with a depth outside the range 2–100, and those with a genotype quality <20 to no call using the SelectVariants command in GATK v. 4.1. Filtered SNPs, indels, sites with >90% missing data, and non-biallelic SNPs were subsequently removed from the callset using bcftools (Danecek et al., 2021).
2.2
Phylogenetic analyses based on mtDNA and nuclear loci
A total of 177 lark sequences were included in the mitochondrial cytb data set (Appendix Table S1). In addition, up to 16 nuclear loci were obtained for 35 taxa (Appendix Table S1). The DNA sequences were trimmed and assembled in MegAlign v. 4.03 in the DNAstar package (DNAstar Inc.). The sequences were aligned using ClustalW (Thompson et al., 1994) implemented in Bioedit 7 (Hall, 1999). The gaps generated by the process of multi-sequence alignments were coded as missing data in all analyses. For each gene, the choice of model was determined based on the Bayesian Information Criterion (BIC) as implemented in jModeltest v. 2 (Darriba et al., 2012). The best-fit models for each dataset are provided in Appendix Table S2.
We used Bayesian inference (BI) to reconstruct a tree from cytb and the 16 nuclear loci for (177 samples), referred to as the 17-locus data. It was analysed using BEAST v. 2.7.1 (Bouckaert et al., 2019), with Panurus biarmicus and Black-chested Prinia (Prinia flavicans) as outgroup (see Appendix Table S1 for details of sampling scheme), using a relaxed lognormal clock model, a birth–death tree prior, and otherwise default priors. Since no suitable primary fossil calibration points are available for Alaudidae, we calibrated the age of the crown clade with a lognormal prior with a standard deviation of 0.08, offset by 19.0. This corresponds to an age of 19 million years (My), with a central 95% prior distribution ranging 16.2–22.2 My, and was based on two independent estimations of the Alaudidae crown node age: (1) multilocus analyses with 13 fossil-calibrated external nodes (~19.0 My ± ~3 My 95% highest posterior distribution (HPD); Stervander et al., 2020b) and (2) analyses of cytb based on a molecular clock (~19.5 My ± ~6 My 95% HPD; Alström et al., 2013).
We ran the Markov Chain Monte Carlo (MCMC) for multiple short rounds of 10–20 million generations, followed by tuning of operators to increase efficiency. We then ran final analyses for 100–125 million generations, sampling every 10,000 generations across eight replicate runs at different starting seeds. Convergence and effective sample size (ESS) was assessed using Tracer v. 1.7.2 (Rambaut et al., 2018), with ESS > 200. For each run, the first 3–5% of the sampled trees were discarded as burn-in, after stationarity had been reached. A maximum credible tree was summarised from the combined replicates with LogCombiner and TreeAnnotator v. 2.7.1 (Rambaut and Drummond, 2015) and displayed in FigTree v. 1.4.3 (Rambaut, 2016).
In addition, a species tree was reconstructed in ASTRAL-III v. 5.7.8 (Zhang et al., 2018) including 177 ingroup samples, with the same outgroup as above. Single-gene trees were inferred using the maximum likelihood (ML) method implemented in IQ-TREE v. 2.0 (Minh et al., 2020) with selection of the best substitution model as implemented in ModelFinder (Kalyaanamoorthy et al., 2017) and branch support measured with the Approximate-Likelihood Ratio Test (alrt). Branches with alrt <80 were collapsed, and the resulting trees were fed into ASTRAL. Branch support in the species tree was measured as local posterior probabilities.
2.3
Phylogenomic SNP data analyses
For phylogenomic analyses we retained 44 samples of 34 species (including the single outgroup species) and added SNP data for 7 samples of 6 Mirafra taxa (M. somalica, M. ashi, M. hypermetra, M. a. africana, M. a. athi, and M. fasciolata) included in Alström et al. (submitted), processed with the above pipeline. We filtered the combined SNPs further to require a minimum genotype quality of 20 (-minGQ 20) and full sample coverage, i.e., none with missing data (-max-missing-count 0) with vcftools v. 0.1.17 (Danecek et al., 2011). Additionally, we excluded SNPs mapped to scaffolds ≤0.5 Mbp, in order to confidently being able to select subsets of physically spaced SNPs. The full final SNP dataset comprised 1,247,784 SNPs across 51 samples of 40 species, referred to as the 51-taxon 1.2M SNP set.
2.3.1
Coalescence-based species tree analyses
We down-sampled the 51-taxon 1.2M SNP set considerably for analyses with SNAPP v. 1.5.1 (Bryant et al., 2012) by (1) retaining a single individual per taxon (41 samples, including the single outgroup species) and (2) thinning by physical distance to include SNPs at a minimum distance of 10 Kbp or larger. From this, we randomly selected two non-overlapping subsets of 5,000 SNPs, creating two 41-taxon 5K SNP sets.
We ran additional analyses with targeted datasets to resolve the branching sequence of (a) the three primary clades and (b) particularly challenging taxa, for which node support was lower in the above analyses (see Results). From the 51-taxon 1.2M SNP set, we selected 21 taxa that represented every genus/major lineage (based on the results from the above analyses) and down-sampled SNPs either (1) through thinning by 50 Kbp, resulting in a matrix of 18,812 SNPs (21-taxon 18K SNP set), or (2) through first thinning by 10 Kbp and then selecting 50,000 of the remaining SNPs at random (21-taxon 50K SNP set). These two datasets addressed the three primary clades and the branching sequence among Calendulauda, Heteromirafra and Mirafra sensu lato. Another dataset specifically addressed the position of Lullula and comprised the seven sampled species within the clade containing Lullula (genera Alauda, Galerida, Lullula, Spizocorys) with Calandrella as outgroup. Thinning by 10 Kbp, we analysed the full matrix of 88,030 variable SNPs (8-taxon 88K SNP set).
Node age calibration was implemented as described for the 17-locus analyses (except in Lullula-specific analyses of the 8-taxon 88K SNP set, in which an uninformative tree age of 10 My was set to improve computation efficiency). We specified that branch-specific population sizes should not be evaluated (through linking theta), using the ruby script snapp_prep.rb (https://github.com/mmatschiner/snapp_prep; Stange et al., 2018). We ran the SNAPP analyses in Beast 2.6.6 (Bouckaert et al., 2019) at different seeds, sampling every 100–500 generations, and optimizing parameters for final runs of typically around 1–2 million generations. We inspected results in Tracer v. 1.7.1 (Rambaut et al., 2018), ensuring stationarity and sufficient effective samples sizes (all parameters >200), and convergence between replicate runs.
2.3.2
Species tree inferred from 50 Kbp windows trees
ASTRAL-III (Zhang et al., 2018) infers a species tree based on individually precomputed trees from discrete loci, such as gene sequences in our multilocus analyses. From the 51-taxon 1.2M SNP set, we extracted all SNPs occurring in 50 Kbp non-overlapping windows at 500 Kbp distance from each other. We retained only those windows containing ≥10 informative sites, resulting in 1370 windows (comprising a total of 67,340 SNPs).
We built individual 50-Kbp-window trees using IQ-TREE v. 2.2.0.3 (Minh et al., 2020) with the same settings and procedures as described above, although we did not explore rate heterogeneity in finer detail. Contracting branches with very low support improves accuracy of ASTRAL (Zhang et al., 2018), hence we used Newick utilities (Junier and Zdobnov, 2010) to remove branches with ultrafast bootstrap support ≤10%, before running ASTRAL-III v. 5.7.8 (Zhang et al., 2018). Since ASTRAL only estimates terminal branch lengths for taxa with multiple samples, we only considered the topology and node support (not branch lengths) of the ASTRAL species trees.
2.3.3
Concatenated ML tree
Using the 51-taxon 1.2M SNP set with IQ-TREE multicore v. 1.6.12 (Nguyen et al., 2015) we implemented ModelFinder (Kalyaanamoorthy et al., 2017) to identify the best substitution model and applied ascertainment bias correction (-m MF + ASC). We then ran additional exploration of rate heterogeneity for the best model GTR + F + ASC + R2 (-m MFP + ASC -cmax 20 -mset GTR + F) that confirmed GTR + F + ASC + R2, and computed a tree with IQ-TREE, obtaining node support with 1000 replicates of ultrafast bootstrap (Hoang et al., 2018).
3.
Results
3.1
Phylogeny based on single-locus and multilocus data
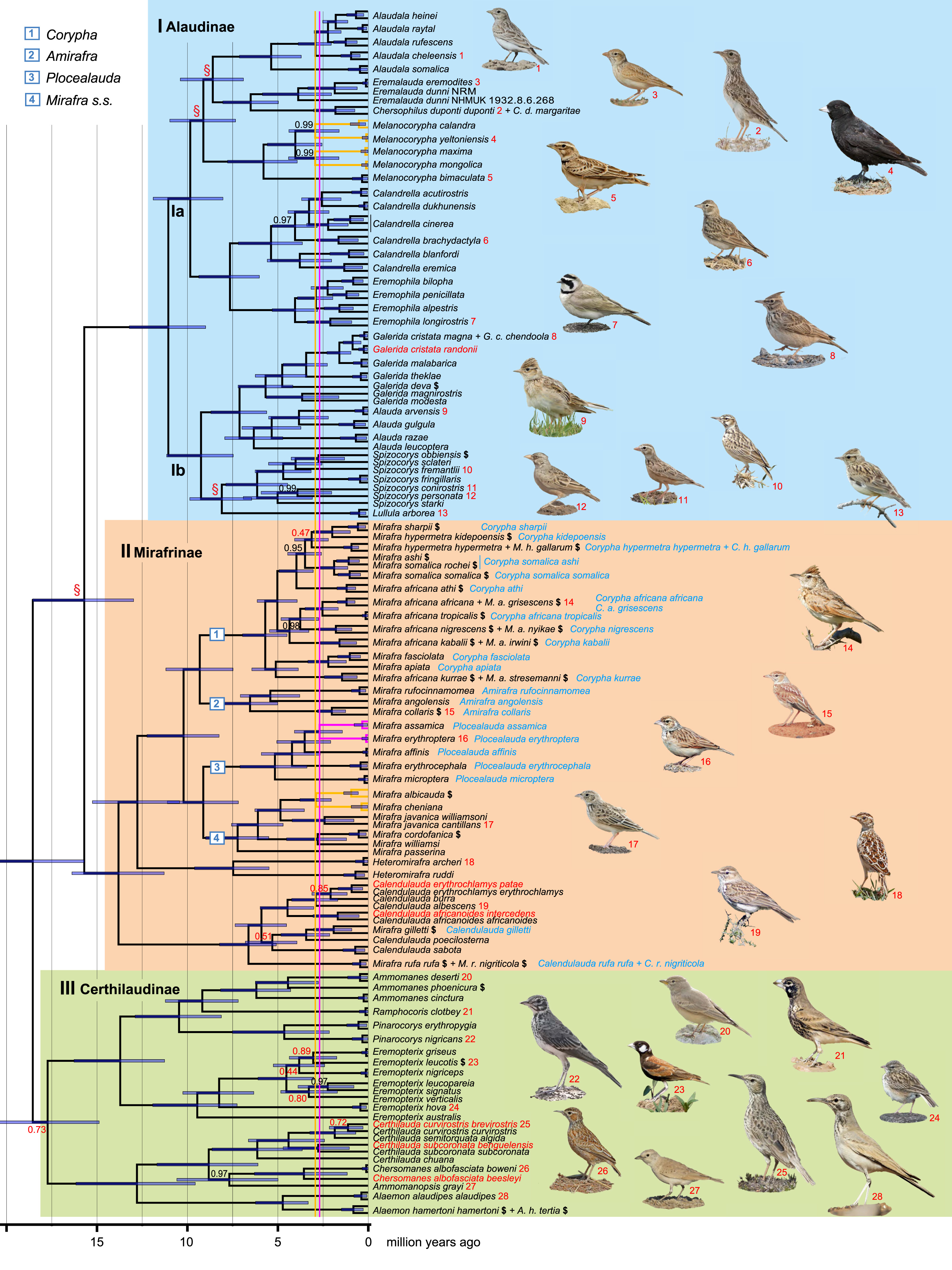
The concatenated 17-locus dataset comprised 177 samples and 14,506 bp, of which 626 sites were variable in mitochondrial cytb and 4,810 sites across the 16 nuclear markers (Appendix Table S2). The tree based on the 17-locus dataset (Fig. 2) includes all of the 93 recognised species in the world (Clements et al., 2022) except Mirafra pulpa. It also covers recently proposed species of Eremophila (Ghorbani et al., 2020b) as well as deeply diverged lineages within Mirafra africana and M. hypermetra, which have been proposed to be treated as separate species (Alström et al.,). In total, 133 taxa are included, of which 44 (including 17 proposed or currently recognised species) are new compared to the study by Alström et al. (2013a). The tree is overall well supported (168 of 176 nodes, 95%, with posterior probability [PP] ≥ 0.95). It shows three primary clades, denoted Ⅰ, Ⅱ and Ⅲ. Clade Ⅰ comprises nine mainly Palearctic genera (with a few species in the Oriental and sub-Saharan African regions) as well as the exclusively sub-Saharan genus Spizocorys. The latter is strongly supported as sister to the Western Palearctic monospecific genus Lullula (Woodlark L. arborea). Clade Ⅱ contains the large genus Mirafra as well as Heteromirafra and Calendulauda. Within Mirafra, all of the species in the clades labelled 1 and 2 are confined to sub-Saharan Africa, all five of the species in clade 3 are endemic to the Oriental region, and five of the seven species in clade 4 are restricted to sub-Saharan Africa, while one (Horsfield’s Bushlark M. javanica) ranges from West Africa to Australia. Two of the Mirafra species (Gillett’s Lark M. gilletti, Rusty Bushlark M. rufa) are part of the Calendulauda clade. Mirafra ashi is weakly differentiated from M. somalica of the subspecies M. s. rochei. Clade III, which is sister to clades Ⅰ and Ⅱ, comprises eight genera, of which half are restricted to sub-Saharan Africa and half occur from southern Africa through the southern part of the Western Palearctic to the Oriental region, and a single species (Madagascar Lark Eremopterix hova) in Madagascar.
Figure
2.
Bayesian (BEAST) multilocus tree of the avian family Alaudidae (larks), based on concatenated mitochondrial cytochrome b and up to 16 nuclear loci per taxon (14,506 bp) analysed in 17 locus-specific partitions. Primary clades discussed in the text have been highlighted and labelled I–III, with subfamily names proposed here. All nodes have posterior probability 1.00, except the ones with values indicated at the nodes (in black when ≥0.95, in red when <0.95). A red § indicates a node that is strongly incongruent with one or more of the SNP-based species trees in Fig. 3. A black $ after a species name indicates that this taxon was not analysed by Alström et al. (2013a). Names in red font are treated as separate species by Gill et al. (2022), see Table 1. The genus Mirafra in clade Ⅱ is proposed to be split into four genera, as indicated by the numbers on the branches and names in the top left corner, to better align with the other genera. The revised taxonomy proposed here is indicated in blue font adjacent to the old names. The four pairs of sympatric sister species with the most recent divergences have been highlighted by yellow/pink branches and vertical lines, to aid comparisons between their divergence times and that of other nodes. The photos show single representatives for all of the genera or, for genera with much variation in structure or plumage (Melanocorypha, Spizocorys, Eremopterix), also deviant species. The names of the photographers are given in the Acknowledgements (in addition: PA number 9–11, 13, 14, 19, 22, 23, and PFD 4–6, 18, 26). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The ASTRAL species tree based on the 17-locus data (Appendix Fig. S1) is less supported than the 17-locus tree based on concatenated sequences (Fig. 2). There are no strongly supported discordances compared to the concatenation tree. The positions of Lullula and Alaudala, which are incongruent among different trees (cf. Figs. 2 and 3), are unresolved. Clades Ⅰ and Ⅱ and the sister relationship between these are strongly supported.
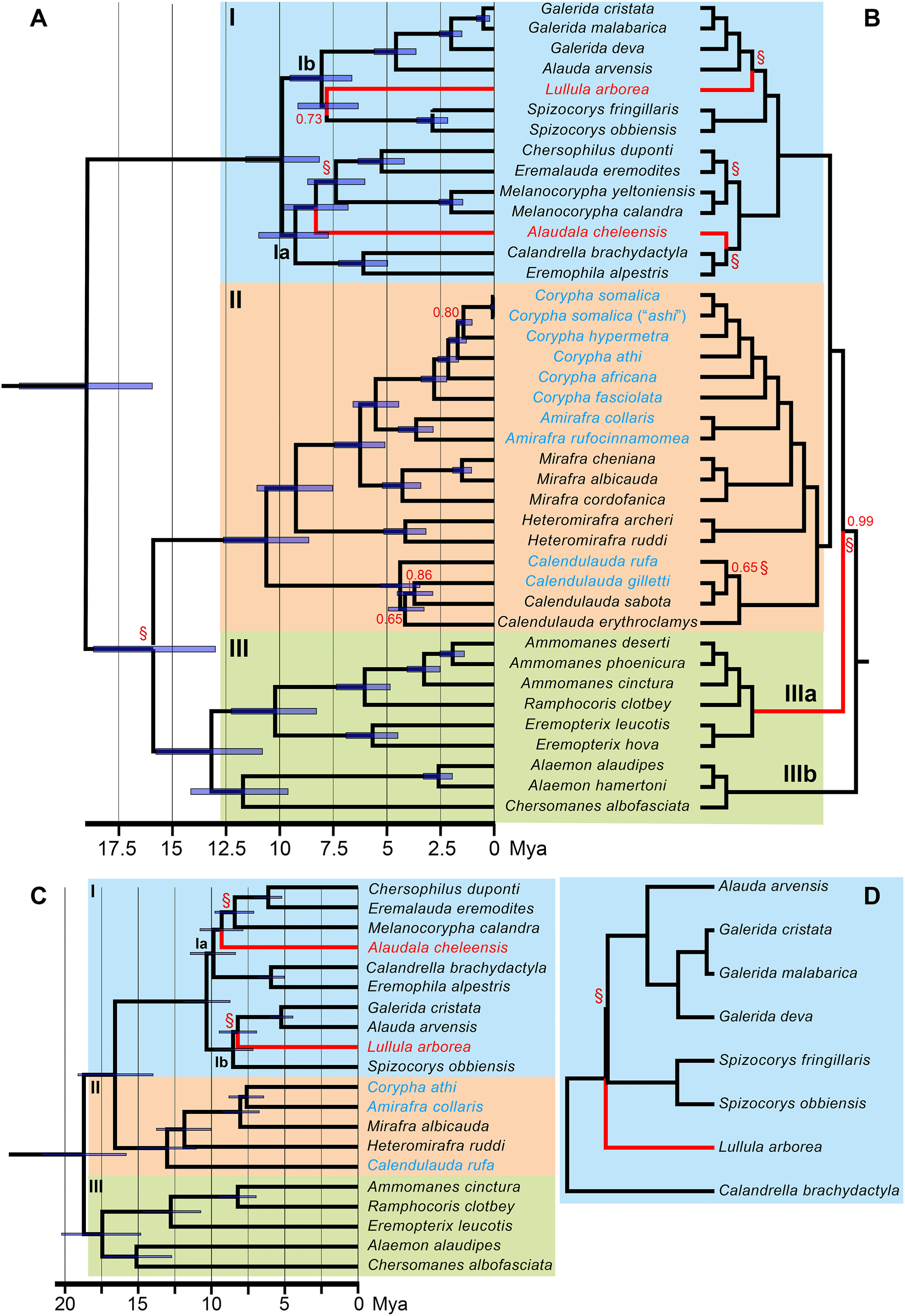
Figure
3.
Bayesian (SNAPP) species tree of the avian family Alaudidae (larks), based on 5,000 genome-wide single nucleotide polymorphisms (SNPs) (A), and ASTRAL species tree, inferred from 1,370 trees, each based on SNPs from well-spaced, non-overlapping 50 Kbp windows (B). The topology of the ASTRAL tree also largely agrees with that of IQ-TREE analyses of all 1,247,784 concatenated SNPs (with all nodes receiving 100% ultrafast bootstrap support; Appendix). Primary clades discussed in the text have been highlighted and labelled I–III (in agreement with Fig. 2). All nodes have posterior probability 1.00, except the ones with values indicated at the nodes. Node bars in the SNAPP tree indicate 95% highest posterior distributions for the divergence times (in the ASTRAL tree, the branch lengths are of fixed length). The red branches are incongruent between these two trees, and strongly supported in at least one of the trees. A red § indicates a strongly supported node that is incongruent with the tree in Fig. 2. The newly proposed names in clade Ⅱ are indicated in blue font (cf. Fig. 2). The two bottom trees are SNAPP species trees based on matrices with a higher number of SNPs and a lower number of taxa, with the aim to resolve the positions of clades Ⅰ, Ⅱ and Ⅲ in relation to each other and the positions of the problematic taxa (Alaudala, Lullula): 50,000 SNPs variable for single representatives per genus from all primary clades (C), and 88,000 SNPs variable for single representatives per genus only from clade Ⅰ (D). The outgroups have been pruned from all trees. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The main SNAPP species trees, based on two non-overlapping 41-taxon 5K SNP datasets and covering representatives of all genera except Pinarocorys, Certhilauda, and Ammomanopsis (Fig. 3A, Appendix Fig. S2), are overall well supported by the data (35–36 of 39 nodes, 90–92%, have PP ≥ 0.95). The two trees only differ on two points: the position of Mirafra hypermetra in relation to M. somalica/M. ashi/M. africana athi, and the position of Lullula, either as sister to Spizocorys (PP = 0.73; Fig. 3A) or to the clade containing Galerida and Alauda (PP = 0.94; Appendix Fig. S2). The two 41-taxon 5K SNP trees generally agree well in topology with the 17-locus tree. However, two incongruences stand out (indicated by red § in Fig. 3A), although only one of these receives PP ≥ 0.95 in both trees: the SNAPP trees recover a sister relationship between clades Ⅱ and Ⅲ whereas the concatenated 17-locus tree recovers a sister relationship between clades Ⅰ and Ⅱ.
The ASTRAL species tree (Fig. 3B) based on >1,300 genomic windows comprising >60,000 SNPs, exhibits even more highly supported nodes (37 of 38 nodes, 97%, have PP ≥ 0.95) than the 41-taxon 5K SNP SNAPP trees. Unlike in the SNAPP analyses, clade Ⅲ is not recovered, but instead is split into two clades (IIIa, IIIb). Five nodes are incongruent with the 17-locus tree (indicated by red § in Fig. 3B). Three of these, concerning the positions of Lullula and Asian Short-toed Lark (Alaudala cheleensis) in clade Ⅰ, and the existence of clade Ⅲ, are incongruent between the SNAPP and ASTRAL trees (indicated by red branches).
The additional SNAPP analyses to further explore these relationships included higher numbers of SNPs: (a) 18,000 or 50,000, respectively, for one species per genus from each of the three primary clades (I–III); and (b) 88,000 SNPs for the species in clade Ib to focus specifically on the position of Lullula. In the 21-taxon 18K SNP (Appendix Fig. S3) and 50K SNP (Fig. 3C) sets, Alaudala remained as sister to the clade comprising Melanocorypha, Chersophilus, and Eremalauda, whereas Lullula moved to the sister position with Galerida and Alauda, and clade Ⅲ was sister to clades Ⅰ and Ⅱ, all with PP = 1.00. In the Lullula-specific analysis (8-taxon 88K SNP; Fig. 3D), Lullula was recovered as sister to Spizocorys, Alauda and Galerida with PP = 1.00. In all these analyses, as well as in the SNAPP tree for all of the species (Fig. 3A), the internode leading up to Lullula and its sister clade was extremely short.
The 51-taxon 1.2M-SNP ML concatenation tree, analysed with IQ-TREE (Appendix Fig. S4), yielded a fully supported topology (100% ultrafast bootstrap support for all nodes) identical to the ASTRAL species tree (Fig. 3B) except for the branching sequence within Calendulauda.
3.3
Divergence time estimates
The node age estimates based on the multilocus (Fig. 2) and genomic data (Fig. 3) differ, with the former being more ancient than the latter (Tables 2 and 3). The youngest estimated mean divergence time for sympatric sister species are 1.1 Mya based on the multilocus data (Mirafra ashi–M. somalica rochei) and 0.06 My based on the SNP data (M. ashi–M. somalica somalica; M. s. rochei not analysed by SNPs), and the second youngest 2.7 My based on the multilocus data (Mirafra assamica–M. erythroptera) and 1.4 My based on the SNP data (Mirafra hypermetra–M. ashi). Two of the taxa that have recently been downgraded from species to subspecies by Clements et al. (2022), namely Chersomanes albofasciata beesleyi and Certhilauda subcoronataa benguelensis, have estimated divergence times from their sister taxon that are older than 2.7 My based on the multilocus data (no SNP data available). The estimated mean divergence times between the newly proposed species range from 2.0 to 5.7 My in the M. africana–M. hypermetra–M. somalica complex (excluding M. ashi; Alström et al., submitted) and from 2.2 to 4.0 My in the Eremophila complex (Table 3).
Table
2.
Divergence time estimates in million years based on the multilocus data (cf. Fig. 2) or, when available, based on the SNP data. Names in red font are treated as separate species by Gill et al. (2022), see Table 1.
Table
3.
Divergence time estimates in million years for the Mirafra africana–M. sharpii–M. hypermetra–M. somalica–M. ashi complex (A) and Eremophila complex (B), based on the multilocus data (cf. Fig. 2) or, when available, based on the SNP data (after a slash symbol; cf. Fig. 3); 95% highest posterior distributions are given in parentheses. Taxa that are believed to be marginally or more widely sympatric are highlighted with blue text (except for M. ashi–M. somalica rochei, which are sympatric, indicated by #; § indicates altitudinal, and hence also habitat, segregation), and taxa that are proposed to be treated as subspecies by Alström et al., (submitted) are highlighted with brown text.
Our analyses of mitochondrial DNA and nuclear introns for all except one of the 93 currently recognised species (Clements et al., 2022) plus several other taxa recently proposed to be distinct species (Drovetski et al., 2014; Ghorbani et al., 2020b; Alström et al., submitted; in total 133 taxa) agrees almost perfectly with the one by Alström et al. (2013a) for the species that were included in both studies. It also recovers strongly supported positions for almost all of the 44 taxa that were not included in the earlier phylogenetic study. Our trees based on genome-wide SNPs added further support to the intergeneric relationships, as well as to previously surprising sister relationships, such as those between Eremalauda and Chersophilus and between Calandrella and Eremophila, as well as Eremopterix hova being far removed from Mirafra (cf. photos in Fig. 2).
All trees in the analyses of the present study are congruent in recovering three primary lineages, except those from SNP-based analyses in ASTRAL and IQ-TREE, which split clade Ⅲ into two non-sister clades (IIIa, IIIb). However, two different topologies reflecting the relationships among the three clades are recovered. Clades II + III are sisters in the SNAPP species trees comprising relatively more species but fewer SNPs (Fig. 3A, Appendix Fig. S2). Conversely, Clades I + II are sisters in the 17-locus tree (Fig. 2) and the two SNAPP trees based on more SNPs and fewer species (Fig. 3C and D, Appendix Fig. S3). The two different topologies receive equally strong support in the respective analyses. The SNAPP species trees (Fig. 3A, D, Appendix Figs. S2–S3) are generally based on a considerably larger number of sites (5,000–50,000 variable sites) than the 17-locus tree (5,436 variable sites, although a significant proportion reflects within-taxon variation in the nuclear markers), and were further analysed under the multispecies coalescent model (MSC; Rannala and Yang, 2003), as was also the ASTRAL tree based on the 17-locus data. The MSC accounts for the ubiquitous heterogeneity in gene trees, thereby providing more realistic support than concatenation for nodes with incongruence among loci or in cases where all or most of the signal in a multilocus analysis comes from a single locus (Degnan et al., 2006; Edwards et al., 2007; Kubatko and Degnan, 2007). However, distinguishing between species tree topologies recovered with fewer sites and more taxa versus more sites and fewer taxa is challenging. We tentatively favour the topology in which clade Ⅲ is sister to clades Ⅰ and Ⅱ, as it is based on a larger number of sites, while acknowledging that sampling more genetic sites does not necessarily lead to more correct trees (Zhang et al., 2021).
The other main incongruences among the trees concern the positions of the monospecific genus Lullula and the genus Alaudala (represented by a single species in the SNP trees, A. cheleensis). In the case of Lullula, there was strong conflict among different analyses (Fig. 3A–D, Appendix Figs. S2 and S3). All of the SNAPP analyses reconstructed a very short internode between Lullula and either of the alternative clades. Such short internodes may be caused either by short time spans between successive lineage splits, which may cause problems for phylogeny reconstruction no matter how much genetic data are analysed, or by ancient introgressive hybridisation, which could also lead to inference of incorrect relationships (Edwards et al., 2016; Taylor and Larson, 2019; Rancilhac et al., 2021; Zhang et al., 2021). While the Eurasian Lullula is sympatric with several Alauda and Galerida species, but widely separated geographically from the Afrotropical Spizocorys, present-day ranges can differ considerably from ancient ones, so Lullula and Spizocorys may have been in geographical contact in the past. At present, the position of Lullula as either sister to the Galerida–Alauda clade or to Spizocorys or to all of these should be considered unresolved. Explicit tests for introgression will be required to test alternative hypotheses (e.g., Green et al., 2010; Burbrink and Gehara, 2018; Zhang et al., 2019; Rancilhac et al., 2021; Zhang et al., 2021).
With respect to Alaudala, all SNAPP analyses support a sister relationship with the Chersophilus–Eremalauda–Melanocorypha clade with full support (Fig. 3A, C, Appendix Figs. S2 and S3). We tentatively support this topology over the alternative one in the ASTRAL and IQ-TREE trees (Fig. 3B; Appendix Fig. S4), where Alaudala is sister to the Calandrella–Eremophila clade. Our preference is based on the fact that SNAPP co-estimates all possible gene trees and directly infers the species tree under the MSC (Bryant et al., 2012). In contrast, ASTRAL infers species trees from precomputed gene trees, while accounting for gene tree discordance under the MSC in a two-step approach (Zhang et al., 2018). The gene trees inferred here by Maximum Likelihood might have estimation errors, which could bias the ASTRAL analysis.
A final incongruence is the position of the Mirafra fasciolata–M. apiata clade. While it is nested within the M. africana–M. sharpii–M. hypermetra–M. somalica–M. ashi complex in the multilocus tree (Fig. 2; not analysed by SNPs in the present study), M. fasciolata is sister to the M. africana–M. sharpii–M. hypermetra–M. somalica–M. ashi complex, with strong support (M. apiata not analysed) in a SNP-based study (Alström et al.,).
4.2
Family level taxonomy
Historically, lark taxonomy has been particularly unstable, owing to a mixture of pronounced convergent and divergent evolution (Alström et al., 2013a), rendering presumed relationships inferred from morphology invalid. However, the vast majority of our analyses – whether based on the 17-locus or SNP data – consistently recovered three well-supported primary clades (I–III), which were also obtained in the more restricted multilocus analyses by Alström et al. (2013a). Given the large number of species within Alaudidae and the frequent historical misclassifications, we suggest that it may be useful for further taxonomic reference to formalise the stable clades Ⅰ–Ⅲ as subfamilies (Table 1), even though no morphological characters consistently distinguish them. We therefore propose:
(1) Subfamily Alaudinae Vigors, 1825 (available under Articles 12.2.4 and 36 of the International Code of Zoological Nomenclature (ICZN, 1999))
Diagnosis: the taxa in this subfamily form a well-supported primary clade in multiple analyses of independent phylogenetic datasets (Fig. 1 in Alström et al., 2013a, Figs. 2 and 3 in the present study), specifically diagnosed by a fixed 1-bp adenine insertion between positions 14 and 15 of Myoglobin (MB) intron 2 in the Zebra Finch reference genome bTaeGut1 v1 (accession GCA_003957565.2); fixed thymine alleles at positions 89 and 434 (other taxa fixed for purines); fixed adenine allele (other taxa fixed for guanine) at position 414 counting from the start of exon 5 of ODC (transcript ODC1-201) in the Zebra Finch reference genome; fixed pyrimidine alleles at position 521 (other taxa fixed for purines).
Included taxa: Alaudinae comprises the Saharo-Arabian genus Eremalauda, the sub-Saharan genus Spizocorys, the Palearctic genera Lullula, Chersophilus and Melanocorypha, and five widespread genera with a predominantly Palearctic distribution: Galerida, Calandrella, Alaudala, Alauda, and Eremophila (all species included in Fig. 2).
(2) Certhilaudinae Le Maout, 1852 (available under Article 11.7.2)
Diagnosis: the taxa in this subfamily form a well-supported primary clade in multiple analyses of independent phylogenetic datasets (Fig. 1 in Alström et al., 2013a, Figs. 2 and 3 in the present study), specifically diagnosed by a fixed guanine allele (adenine in all other taxa) at position 19 and a fixed cytosine allele (guanine in all other taxa; site not sampled in Pinarocorys) at position 665 in Myoglobin; reference and positions as declared above for Alaudinae.
Included taxa: Certhilaudinae comprises the sub-Saharan genera Ammomanopsis, Chersomanes, Certhilauda, and Pinarocorys, the predominantly Saharo-Arabian genera Alaemon, Ramphocoris, and Ammomanes, and the Afro-Oriental genus Eremopterix (all species included in Fig. 2).
(3) Mirafrinae Bianchi, 1905 (available under Article 12.2.4)
Diagnosis: the taxa in this subfamily form a well-supported primary clade in multiple analyses of independent phylogenetic datasets (Fig. 1 in Alström et al., 2013a, Figs. 2 and 3 in the present study), specifically diagnosed by a fixed guanine allele (adenine in all other taxa) at position 544 in Myoglobin and a fixed adenine allele or deletion (the latter only in Corypha hypermetra; all other taxa fixed for guanine) at position 534 of ODC; reference and positions as declared above for Alaudinae.
Included taxa: Mirafrinae comprises the exclusively sub-Saharan genera Calendulauda, Heteromirafra, Amirafra, and Corypha, the predominantly sub-Saharan genus Mirafra, and the Oriental genus Plocealauda (all species except Mirafra pulpa are included in Fig. 2).
We follow the principles of a linear phylogenetic sequence employed by Fjeldså et al. (2020b) to order the subfamilies, genera, and species (Table 1), tentatively placing more weight on the large-scale phylogenomic and multilocus results that recover Alaudinae (clade Ⅰ) and Mirafrinae (clade Ⅱ) as sisters (Fig. 3).
4.3
Genus level taxonomy
The genus Mirafra is considerably larger than any of the other genera, with deep internal divergences that are considerably older than several of the other genera in the family. In order to create a more balanced taxonomy, we propose to split the present Mirafra into four genera, corresponding to the four primary clades within Mirafra (indicated by 1–4 in Fig. 2). Names are available for three: Mirafra sensu stricto, i.e., MirafraHorsfield, 1821 (type M. javanica), CoryphaG.R. Gray, 1840 (type C. apiata), and AmirafraBianchi, 1906 (type A. collaris). For the fourth clade, PlocealaudaHodgson in J.E. Gray, 1844 has been used. However, this is a nomen nudum and therefore unavailable. As no other names are available for this clade, and with the intention not to inflate the number of names further, we here formally describe:
Genus Plocealauda gen. nov.
Type species (by designation): Plocealauda assamica (Horsfield, 1840).
Diagnosis: The Plocealauda larks have a rather thick-set appearance, with heavy bills, nostrils not covered by feathers, moderately long hind claws, and short, rounded wings with primaries not projecting or just projecting slightly beyond the tips of the tertials on the folded wings. They have well developed outermost primary, rounded (not markedly notched) tips to the secondaries and inner primaries, three tertials, and relatively short tails. They lack distinct crests, although one species regularly raises its crown feathers, and are heavily streaked above and on the breast, with conspicuous rufous panels on the primaries. None of these morphological characters are truly diagnostic; however, the species in this genus form a well-supported clade in multilocus analyses (Fig. 1 in Alström et al., 2013a, Fig. 2 in the present study) as well as single-locus analyses of cytb, Myoglobin, and ODC (not shown here). The genus is specifically diagnosed by a fixed cytosine allele (adenine in all other taxa) at position 642 of cytb, a fixed cytosine allele (thymine/adenine in all other taxa) at position 424 of ODC, and fixed alleles at five positions of Myoglobin: 99 (thymine vs. guanine in all other taxa), 177 (adenosine vs. cytosine/deletion), 324 (thymine vs. cytosine), 332 (cytosine vs. adenosine/guanine), and 578 (cytosine vs. adenosine); reference and positions as declared above for Alaudinae.
Included species: Plocealauda comprises P. microptera, P. erythrocephala, P. affinis, P. erythroptera, and P. assamica; all species are monotypic and their distributions range from the Indian Subcontinent to Southeast Asia.
Nomenclatural acts: The LSID for the nomenclatural act associated with Plocealauda is urn:lsid:zoobank.org:act:3EB6ACAE-CA27-4340-831B-BECFFBCFEDFE. The LSID for this publication is urn:lsid:zoobank.org:pub:4E231C1E-BCD2-4580-920E-A83F2E0F93D1. The electronic edition of this work was published in a journal with ISSN 2053–7166, and is available from the digital repository http://www.ncbi.nlm.nih.gov/pmc/. The electronic edition of this article conforms to the requirements of Article 8.5.3 of the amended International Code of Zoological Nomenclature (ICZN, 2012), and hence the new name contained herein are available under that Code from the electronic edition of this article. This published work and the nomenclatural acts it contains have been registered in ZooBank, the online registration system for the International Commission of Zoological Nomenclature. The ZooBank Life Science Identifiers (LSIDs) can be resolved and the associated information viewed through any standard web browser by appending the LSID to the prefix “http://zoobank.org/”.
Mirafra sensu stricto is the only genus that is widespread (West Africa to Australia, although only one of the species occurs outside of sub-Saharan Africa). We tentatively place M. pulpa in this genus, as it is very similar to the other species, especially to M. passerina in song and other behaviours (Bradley, 2021). Both Corypha and Amirafra are restricted to sub-Saharan Africa, while Plocealauda is confined to South and Southeast Asia.
Based on our results, the previously phylogenetically unstudied Mirafra gilletti and M. rufa are moved to Calendulauda, i.e., Calendulauda gilletti and C. rufa, respectively. With regard to other previously unsampled species, the oddly-plumaged Collared Lark (Mirafra collaris) is placed in the revived genus Amirafra, Kordofan Lark (Mirafra cordofanica) is determined to belong in Mirafra sensu stricto, Obbia Lark (Spizocorys obbiensis) is confirmed as being part of the genus Spizocorys, and Lesser Hoopoe-lark (Alaemon hamertoni) is confirmed to be sister to Greater Hoopoe-lark (A. alaudipes).
4.4
Species level taxonomy
The Corypha (i.e., until now Mirafra) africana–C. sharpii–C. hypermetra–C. somalica–C. ashi complex has recently been revised using an integrative taxonomic approach (Alström et al., submitted). That study proposed that nine species should be recognised in this complex: C. somalica (with C. ashi treated as a synonym of C. somalica rochei), C. hypermetra, C. sharpii, C. kidepoensis, C. athi, C. africana sensu stricto, C. nigrescens, C. kabalii and C. kurrae. Corypha sharpii has recently (October 2022) been recognised as a monotypic species (Sharpe’s Lark Mirafra sharpii; Clements et al., 2022; Alström and Donald, 2022). The present study supports the taxonomy proposed by Alström et al. (submitted) (Fig. 2). First, all of the traditional species (except M. ashi, which is monotypic) are recovered as paraphyletic. Second, the youngest divergence times based on the multilocus data between taxa that are usually treated as subspecies of M. africana and the other currently recognised species in this complex are estimated at 2.0 My (M. sharpii vs. M. hypermetra kidepoensis) and 4.0 My (M. africana athi vs. the other species) (Table 2). Moreover, the estimated mean divergence times among the M. africana subspecies that were proposed to be treated as separate species by Alström et al. (submitted) are 4.0–5.7 My (Table 2). These divergence times are older than or equal to those between most of the sympatric lark species pairs (Table 2). In addition, some of the youngest divergences in this complex concern taxa that are considered to be sympatric: M. hypermetra hypermetra and M. somalica rochei + M. ashi, and M. s. somalica and M. sharpii (Ash and Miskell, 1998). With respect to M. ashi, the multilocus analysis indicates 1.1 My divergence from its sister taxon M. somalica rochei, whereas the differentiation between M. ashi and M. s. somalica was only 0.06 My according to the SNP analysis (M. s. rochei not analysed by SNPs). Alström et al. (submitted) concluded, based on an integrative taxonomic approach, that rochei should be synonymised with ashi (by priority).
The genus Eremophila has been suggested to be treated as six species (Drovetski et al., 2014) or as four species (Ghorbani et al., 2020b) based mainly on mitochondrial DNA. The present study supports recognition of at least four species, as the youngest sister pair, E. bilopha–E. penicillata, are locally sympatric, although segregated by elevation and habitat (Cramp, 1988; Shirihai and Svensson, 2018). The estimated divergence times among the other proposed Eremophila species are on par with those between some other sympatric lark species pairs (Table 2). Genomic data would be valuable for further confirmation.
Galerida malabarica has previously only been analysed by a short cytb (550 bp) and Fib7 (115 bp) sequences (Guillaumet et al., 2008; Alström et al., 2013a). In the 17-locus analysis it is found to be sister to the pair composed of G. cristata (G. c. magna and G. c chendoola) and the recently lumped Maghreb Lark G. cristata randonii (Clements et al., 2022; treated as G. macrorhyncha by Gill et al., 2022), with an estimated divergence time that is younger than any of the sympatric species (Table 2). It is by far the youngest of the sister pairs analysed with SNP data, with a divergence time of only one third of the youngest sympatric species pair (Table 2). Future studies are needed to determine whether these two Galerida species, which are essentially parapatrically distributed but with an apparent narrow zone of sympatry (Ganpule et al., 2022), are reproductively isolated or should rather be treated as conspecific. Galerida cristata magna/G. c chendoola and G. c. randonii are even more shallowly diverged (Table 2), and are treated as conspecific by one of the main checklists (Clements et al., 2022). However, they have shown evidence of reproductive isolation in a contact zone in Morocco (Guillaumet et al., 2005, 2006, 2008).
The other shallowly diverged para-/allopatrically distributed species pairs that have recently been lumped (Clements et al., 2022) have variously deep divergence times (Table 2): Beesley’s Lark (Chersomanes a. beesleyi)–Spike-heeled Lark (C. albofasciata), Agulhas Long-billed Lark (Certhilauda curvirostris brevirostris)–Cape Long-billed Lark (C. curvirostris), Benguela Long-billed Lark (C. subcoronata benguelensis)–Karoo Lark (C. subcoronata), Foxy Lark (Calendulauda africanoides intercedens, formerly C. alopex intercedens)–Fawn-coloured Lark (C. africanoides), Barlow’s Lark (C. erythrochlamys patae, formerly C. barlowi patae)–Dune Lark (C. erythrochlamys), and Singing Bushlark (M. javanica cantillans)–M. javanica. Although three of these six pairs have more recent divergence times (0.9–1.8 My) than any of the sympatric species pairs, three of them (C. a. beesleyi–C. albofasciata, C. s. benguelensis–C. subcoronata and Mirafra j. cantillans–M. javanica) have estimated divergence times (3.8 My, 2.8 My and 2.4 My, respectively) that overlap those of several sympatric species pairs. Moreover, if C. curvirostris and C. brevirostris are treated as conspecific, Eastern Long-billed Lark (C. semitorquata) should perhaps be included in that species, as it is only slightly more diverged (Table 2). As these analyses are based mainly or exclusively on mitochondrial DNA (cf. Appendix Table S1), more comprehensive analyses are warranted to evaluate the taxonomic status of these taxa.
5.
Conclusions
Our study infers a baseline phylogeny of the species-rich lark family based on multilocus sequence data combined with up to 1.2 million SNPs. It confirms the previously most densely sampled study, which revealed an unusually poor correspondence between phenotypes and phylogenetic relationships. We define three primary clades as subfamilies and balance the taxonomy of the family by splitting the large genus Mirafra into four genera, with divergence times similar to those between other lark genera. Two species are moved from Mirafra to Calendulauda, and several subspecies in a Corypha (formerly Mirafra) species complex that were suggested to be raised to species rank in a different study are here supported as separate species.
This updated phylogeny and taxonomy of Alaudidae will serve as a foundation for future studies of various aspects of lark evolution and natural history, including addressing the few remaining phylogenetic conflicts uncovered here. Our findings further highlight the need for exploring biogeography, species delimitation, and systematics of the widespread and subspecies-rich Ammomanes and Alauda larks, as well as several understudied sub-Saharan taxa.
Authors’ contributions
PA and PFD conceived the study; PA, DE, PAC, AG and BIT collected key samples; MI, UO and PA did the lab work for whole-genome sequencing; EE and MS conducted the bioinformatic analyses; MS performed all phylogenomic analyses; ZM performed alignments; MS did the multilocus and ZM and LR the single-locus phylogenetic analyses; LR did the ASTRAL analyses of the multilocus data; PA and MS wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Ethics statement
Much research in this paper is based on pre-existing museum collections that have been amassed over many decades, however strictly regulated only during later years. Modern field studies and collection contributing to this study comply with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora. Field work and sample collection in Morocco was specifically approved under permit 4177 MCEF/CRF/CPB/PFF. Field work and sample collection in Kenya did not involve endangered or protected species and was approved by The National Museums of Kenya. Field work and sample collection in Saudi Arabia was approved by HH Prince Bandar bin Saud, president of the Saudi Wildlife Commission, and Mr Ahmad Al Bouq, director of the National Wildlife Research Center.
Declaration of competing interest
As authors of manuscript Systematics of the avian family Alaudidae using multilocus and genomic data, we declare that we have no financial or personal relationships that influence our work.
Acknowledgements
We are indebted to Mark Adams and Hein van Grouw and the Natural History Museum, Tring, UK, for access to specimens and multiple toepad samples for DNA; to Sharon Birks and the University of Washington Burke Museum, David Willard and the Field Museum, Chicago, Sylke Frahnert and the Museum Für Naturkunde, Berlin, Nigel Collar and Claire Spottiswoode for DNA samples; to Nick Horrocks for managing samples from Kenya; Robert Dowsett and David Donsker for comments on nomenclature; to the National Swedish Research Council (grants No. 2015-04402, 2019-04486), the Carl Trygger Foundation (CTS 20: 6), the Jornvall Foundation, and Julian Francis for financial support; and to the National Genomics Infrastructure in Stockholm funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation; the Swedish Research Council, and the Swedish National Infrastructure for Computing (SNIC)/Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure, and SciLifeLab for next-generation DNA sequencing; and to the Research/Scientific Computing teams at The James Hutton Institute and NIAB for providing computational resources and technical support for the “UK’s Crop Diversity Bioinformatics HPC” (BBSRC grant BB/S019669/1). We are grateful to Mansur al Fahad (3), Klas Alström (7), Hadi Ansari (20), Nik Borrow (15), Tomas Carlberg (12, 17), Göran Ekström (14), David Erterius (24), the late Joe Grosel (25, 27), Zongzhuang Liu (8), Vincent Legrand (2), Jonathan Martínez (1), Magnus Ullman (28) and Arnoud van den Berg (21) for the use of their photos in Fig. 2 (the numbers refer to the labels on the photos), and to Beatriz Acuña Hidalgo for editing of these photos. We are indebted to Richard L. Pyle for exemplary help with registration of nomenclatural acts. We thank the editor and two anonymous reviewers for comments on a previous version of the manuscript. Laurent Raty and Jimmy Gaudin keenly spotted nomenclatural issues that we could thereby address before the publication of the final version of this paper, with additional research and invaluably insightful input from Richard Schodde, Frank Rheindt, Laurent Raty, and Alan Peterson.
Appendix A. Supplementary data
The 700 sequences generated in this study have been submitted to GenBank under the accession numbers OQ550976–OQ551097 and OQ595424–OQ596001 (see Appendix Table S1). SNP datasets (vcf files) and tree output have been deposited to Zenodo, with the https://doi.org/10.5281/zenodo.7643432
Ali, S., Ripley, S.D., 1973. Handbook of the Birds of India and Pakistan. vol. 9. Oxford University Press, Bombay.
Alström, P., 1998. Taxonomy of the Mirafra assamica complex. Forktail 13, 97-107.
Alström, P., 2020a. Tibetan Lark (Melanocorypha maxima), version 1.0. In: del Hoyo, J., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E. (Eds.), Birds of the World. Cornell Lab of Ornithology, Ithaca. .
Alström, P., 2020b. Mongolian Lark (Melanocorypha mongolica), version 1.0. In: del Hoyo, J., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E. (Eds.), Birds of the World. Cornell Lab of Ornithology, Ithaca. .
Alström, P., Donald, P.F., 2022. Sharpe’s Lark (Mirafra sharpii), version 1.0. In: Keeney, B.K. (Ed.), Birds of the World. Cornell Lab of Ornithology, Ithaca. .
Alström, P., Sundev, G., 2021. Mongolian Short-toed Lark Calandrella dukhunensis, an overlooked East Asian species. J. Ornithol. 162, 165-177. .
Alström, P., Mohammadi, Z., Donald, P.F., Nymark, M., Enbody, E.D., Irestedt, M., et al. Integrative Taxonomy Reveals Unrecognised Species Diversity in African Corypha Larks (Aves: Alaudidae). (Submitted manuscript).
Alström, P., Ericson, P.G.P., Olsson, U., Sundberg, P., 2006. Phylogeny and classification of the avian superfamily Sylvioidea. Mol. Phylogenet. Evol. 38, 381-397. .
Alström, P., Barnes, K.N., Olsson, U., Barker, F.K., Bloomer, P., Khan, A.A., et al., 2013a. Multilocus phylogeny of the avian family Alaudidae (larks) reveals complex morphological evolution, non-monophyletic genera and hidden species diversity. Mol. Phylogenet. Evol. 69, 1043-1056. .
Alström, P., Olsson, U., Lei, F., 2013b. A review of the recent advances in the systematics of the avian superfamily Sylvioidea. Chinese Birds 4, 99-131. .
Alström, P., van Linschooten, J., Donald, P.F., Sundev, G., Mohammadi, Z., Ghorbani, F., et al., 2021. Multiple species delimitation approaches applied to the avian lark genus Alaudala. Mol. Phylogenet. Evol. 154, 106994. .
Andrews, S., 2010. FastQC: a quality control tool for high throughput sequence data. .
Bianchi, V.L., 1905. Дополнительныя замітки о палеарктическихь жаворонкахъ (Alaudidae) [Notices supplémentaires sur les alouettes paléarctiques (Alaudidae)]. Bulletin de l’Académie impériale des sciences de St.-Pétersbourg XXIII, 205–240, 5esérie.
Bianchi, V., 1906. Catalogue of the known species of Alaudidae or family of Larks, with a
Table showing the geographical distribution and a Key to the genera. Bulletin de
l’Académie impériale des sciences de St.-Pétersbourg. 5e série XXV, 1–98.
Bouckaert, R., Vaughan, T.G., Barido-Sottani, J., Duchene, S., Fourment, M., Gavryushkina, A., et al., 2019. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650. .
Bradley, J., 2021. Friedmann’s Lark (Mirafra pulpa), version 2.0. In: Kirwan, G.M., Keeney, B.K. (Eds.), Birds of the World. Cornell Lab of Ornithology, Ithaca. .
Bryant, D., Bouckaert, R., Felsenstein, J., Rosenberg, N.A., RoyChoudhury, A., 2012. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917-1932.
Burbrink, F.T., Gehara, M., 2018. The biogeography of deep time phylogenetic reticulation. Syst. Biol. 67, 743-755.
Clements, J.F., Schulenberg, T.S., Iliff, M.J., Fredericks, T.A., Gerbracht, J.A., Lepage, D., et al., 2022. The eBird/Clements checklist of Birds of the World: v2022. .
Cramp, S., 1988. In: The Birds of the Western Palearctic, vol. V. Oxford University Press, Oxford.
Danecek, P., Auton, A., Abecasis, G., Albers, C.A., Banks, E., DePristo, M.A., et al., 2011. 1000 Genomes Project Analysis Group, The variant call format and VCFtools. Bioinformatics 27, 2156-2158.
Danecek, P., Bonfield, J.K., Liddle, J., Marshall, J., Ohan, V., Pollard, M.O., et al., 2021. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. .
Darriba, D., Taboada, G.L., Doallo, R., Posada, D., 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat. Method. 9, 772. .
de Juana, E., Suárez, F., Ryan, P., Alstrom, P., Donald, P., 2004. Family Alaudidae (larks).
In: del Hoyo, J., Elliott, A., Christie, D.A. (Eds.), Handbook of the Birds of the World,
vol. 9. Lynx Edicions, Barcelona, pp. 496–601.
Degnan, J.H., Rosenberg, N.A., 2006. Discordance of species trees with their most likely gene trees. PLoS Genet. 2, 762-768.
Dement’ev, G.P., Gladkov, N.A., 1968. Birds of the Soviet Union, vol. 5. Israel Program for
Scientific Translations.
Donald, P.F., Christodoulides, S., 2018. The 2007 record of ‘Dunn’s Lark’ on Cyprus revisited, with notes on the separation of Dunn’s Lark Eremalauda dunni and Arabian Lark E. eremodites. Sandgrouse 40, 17-24.
Donald, P.F., Collar, N.J., 2011. Notes on the structure and plumage of Beesley’s Lark Chersomanes [albofasciata] beesleyi. Bull. Afr. Bird Club 18, 168-173.
Donald, P., Alström, P., Engelbrecht, D., 2017. Possible mechanisms of substrate colour-matching in larks (Alaudidae) and their taxonomic implications. Ibis 159, 699-702. .
Drovetski, S.V., Raković, M., Semenov, G., Fadeev, I.V., Red’kin, Y.A., 2014. Limited phylogeographic signal in sex-linked and autosomal loci despite geographically, ecologically, and phenotypically concordant structure of mtDNA variation in the Holarctic avian genus Eremophila. PLoS One 9, e87570. .
Edwards, S.V., Liu, L., Pearl, D.K., 2007. High-resolution species trees without concatenation. P. Natl. Acad. Sci. USA 104, 5936-5941.
Edwards, S.V., Potter, S., Schmitt, C.J., Bragg, J.G., Moritz, C., 2016. Reticulation, divergence, and the phylogeography-phylogenetics continuum. P. Natl. Acad. Sci. USA 113, 8025-8032.
Finch, B.W., Hatfield, R.S., Colombo, S., Kennedy, A.S., te Raa, M., Irestedt, M., et al., 2023. Disjunct resident population of Melodious Lark Mirafra cheniana discovered in East Africa. J. Ornithol. 164, 55-71. .
Fjeldså, J., Alström, P., Olsson, U., Cibois, A., Johansson, U., 2020a. Superfamily Sylvioidea, the old world warblers and their allies. In: Fjeldså, J., Ericson, P.G.P., Christidis, L. (Eds.), The Largest Avian Radiation: the Evolution of Perching Birds, or the Order Passeriformes. Lynx Edicions, Barcelona, pp. 191–236.
Fjeldså, J., Christidis, L., Ericson, P.G.P., Stervander, M., Ohlson, J.I., Alström, P., 2020b. An updated classification of passerine birds. In: Fjeldså, J., Ericson, P.G.P., Christidis, L. (Eds.), The Largest Avian Radiation: the Evolution of Perching Birds, or the Order Passeriformes. Lynx Edicions, Barcelona, pp. 45–64.
Freed, D., Aldana, R., Weber, J.A., Edwards, J.S., 2017. The Sentieon Genomics Tools - A fast and accurate solution to variant calling from next-generation sequence data. BioRxiv, 115717. .
Fregin, S., Haase, M., Olsson, U., Alström, P., 2012. New insights into family relationships within the avian superfamily Sylvioidea (Passeriformes) based on seven molecular markers. BMC Evol. Biol. 12, 157. .
Ganpule, P., Varu, M., Trivedi, B., Raina, A.D., 2022. A Field Guide to the Birds of Gujarat.
Bird Conservation Society, Gujarat, Ahmedabad.
Ghorbani, F., Aliabadian, M., Zhang, R., Irestedt, M., Yan, H., Sundev, G., et al., 2020a. Densely sampled phylogenetic analyses of the Lesser Short-toed Lark Alaudala rufescens-Sand Lark A. raytal species complex (Aves, Passeriformes) reveal cryptic diversity. Zool. Scripta 49, 427-439. .
Ghorbani, F., Aliabadian, M., Olsson, U., Donald, P.F., Khan, A.A., Alström, P., 2020b. Mitochondrial phylogeography of the genus Eremophila confirms underestimated species diversity in the Palearctic. J. Ornithol. 161, 297-312. .
Gill, F., Donsker, D., Rasmussen, P., 2022. IOC world bird list (v12.2). .
Gray, G.R., 1840. A List of the Genera of Birds, with an Indication of the Typical Species of
Each Genus. Richard and John E. Taylor, London.
Green, R.E., Krause, J., Briggs, A.W., Maricic, T., Stenzel, U., Kircher, M., et al., 2010. A draft sequence of the Neandertal genome. Science 328, 710-722.
Guillaumet, A., Crochet, P.A., Godelle, B., 2005. Phenotypic variation in Galerida larks in Morocco: the role of history and natural selection. Mol. Ecol. 14, 3809-3821.
Guillaumet, A., Pons, J.M., Godelle, B., Crochet, P.A., 2006. History of the Crested Lark in the Mediterranean region as revealed by mtDNA sequences and morphology. Mol. Phylogenet. Evol. 39, 645-656.
Guillaumet, A., Crochet, P.A., Pons, J-M., 2008. Climate-driven diversification in two widespread Galerida larks. BMC Evol. Biol. 8, 32.
Hall, T.A., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95⁄98⁄NT. Nucleic Acids Symp. Ser. 41, 95-98.
Hoang, D.T., Chernomor, O., von Haeseler, A., Minh, B.Q., Vinh, L.S., 2018. UFBoot2: improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518-522.
Hodgson, B.H., 1844. Catalogue of Nipalese birds, collected between 1824 and 1844. Zoological Miscellany (J.E. Gray), 81–86.
Horsfield, T., 1821. Systematic arrangement and description of birds from the island of Java. Trans. Linn. Soc. London 13, 133–200.
Horsfield, T., 1840. List of Mammalia and Birds collected in Assam by John McClelland, Esq., Assistant-Surgeon in the service of the East India Company, Bengal Establishment, Member of the late deputation which was sent into that country for the purpose of investigating the nature of the Tea Plant. Proc. Zool. Soc. Lond. 7, 146–167.
ICZN (International Commission on Zoological Nomenclature), 1999. International Code
of Zoological Nomenclature, fourth ed. International Trust for Zoological
Nomenclature, London, p. 335.
ICZN (International Commission on Zoological Nomenclature), 2012. Amendment of Articles 8, 9, 10, 21 and 78 of the International Code of Zoological Nomenclature to Expand and Refine Methods of Publication. ZooKeys 219, 1-10. .
Irestedt, M., Thorn, F., Muller, I., Joensson, K.A., Ericson, P.G.P., 2022. A guide to avian museomics: insights gained from resequencing hundreds of avian study skins. Mol. Ecol. Res. 22, 2672-2684. .
Junier, T., Zdobnov, E.M., 2010. The Newick utilities: high-throughput phylogenetic tree processing in the UNIX shell. Bioinformatics 29, 1669-1670.
Kalyaanamoorthy, S., Minh, B.Q., Wong, T.K.F., von Haeseler, A., Jermiin, L.S., 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Method. 14, 587-589. .
Kubatko, L.S., Degnan, J.H., 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56, 17-24.
Le Maout, J.E.M., 1852. Histoire naturelle des oiseaux: suivant la classification de M.
Isidore Geoffroy-Saint-Hilaire, avec l’indication de leurs moeurs et de leurs rapports
avec les arts, le commerce et l’agriculture. L. Curmer, Paris.
Li, H., 2013. Aligning sequence reads, clone sequences and assembly contigs with BWAMEM. arXiv, 1303.3997. .
Linnaeus, C., 1758. Systema Naturae per regna tria naturae, secundum classes, ordines,
genera, species, cum characteribus, differentiis, synonymis, locis. Editio decima,
reformata, Vol. 1. Laurentius Salvius, Holmiae.
MMason, N.A., Pulgarin, P., Cadena, C.D., Lovette, I.J., 2020. De novo assembly of a high-quality reference genome for the Horned Lark (Eremophila alpestris). G3: Genes Genomes Genet. 10, 475–478. .
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al., 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297-1303. .
Meyer, M., Kircher, M., 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocals 2010(6), 1-10. .
Minh, B.Q., Schmidt, H.A., Chernomor, O., Schrempf, D., Woodhams, M.D., von Haeseler, A., et al., 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530-1534. .
Nguyen, L.-T., Schmidt, H.A., von Haeseler, A., Minh, B.Q., 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268-274.
Oliveros, C.H., Field, D.J., Ksepka, D.T., Barker, F.K., Aleixo, A., Andersen, M.J., et al., 2019. Earth history and the passerine superradiation. P. Nat. Acad. Sci. USA 116, 7916-7925. .
Peters, J.L., 1960. Family Alaudidae. In: Mayr, E., Greenway Jr., J.C. (Eds.), Check-list of
Birds of the World, vol. IX. Museum of Comparative Zoology, pp. 3–80. Cambridge,
Mass.
Rambaut, A., 2016. FigTree. Version 1.4.3. .
(Accessed 4 October 2016).
Rambaut, A., Drummond, A.J., 2015. TreeAnnotator. Version 2.2.1. .
Rambaut, A., Drummond, A.J., Xie, D., Baele, G., Suchard, M.A., 2018. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901-904. .
Rancilhac, L., Irisarri, I., Angelini, C., Arntzen, J.W., Babik, W., Bossuyt, F., et al., 2021. Phylotranscriptomic evidence for pervasive ancient hybridization among Old World salamanders. Mol. Phylogenet. Evol. 155, 106967. .
Rannala, B., Yang, Z., 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164, 1645-1656.
Rasmussen, P.C., Anderton, J.C., 2012. In: Birds of South Asia: the Ripley Guide, 2nd
revised edition, vol. 2. Smithsonian Institution and Michigan State University, Lynx
Edicions, Barcelona.
Roberts, T.J., 1992. In: The Birds of Pakistan, vol. 2. Oxford University Press, Karachi.
Ryan, P.G., Bloomer, P., 1999. The Long-billed Lark complex: a species mosaic in south-western Africa. Auk 116, 194-208.
Ryan, P.G., Hood, I., Bloomer, P., Komen, J., Crowe, T.M., 1998. Barlow’s Lark: a new species in the Karoo Lark Certhilauda albescens complex of southwest Africa. Ibis 140, 605-619.
Shirihai, H., Svensson, L., 2018. In: Handbook of Western Palearctic Birds, vol. 1.
Passerines to Larks. Helm, London.
Sigeman, H., Ponnikas, S., Hansson, B., 2020. Whole-genome analysis across 10 songbird families within Sylvioidea reveals a novel autosome-sex chromosome fusion. Biol. Lett. 16, 20200082. .
Spottiswoode, C.N., Olsson, U., Mills, M.S.L., Cohen, C., Francis, J.E., Toye, N., et al., 2013. Rediscovery of a long-lost lark reveals the conspecificity of endangered Heteromirafra populations in the Horn of Africa. J. Ornithol. 154, 813-825. .
Stange, M., Sanchez-Villagra, M.R., Salzburger, W., Matschiner, M., 2018. Divergence-time estimation with genome-wide single-nucleotide polymorphism data of sea catfishes (Ariidae) supports miocene closure of the Panamanian Isthmus. Syst. Biol. 67, 681-699.
Stervander, M., Alström, P., Olsson, U., Ottosson, U., Hansson, B., Bensch, S., 2016. Multiple instances of paraphyletic species and cryptic taxa revealed by mitochondrial and nuclear RAD data for Calandrella larks (Aves: Alaudidae). Mol. Phylogenet. Evol. 102, 233-245. .
Stervander, M., Hansson, B., Olsson, U., Hulme, M.F., Ottosson, U., Alström, P., 2020a. Molecular species delimitation of larks (Aves: Alaudidae), and integrative taxonomy of the genus Calandrella, with the description of a range-restricted African relic taxon. Diversity 12, 428. .
Stervander, M., Fjeldså, J., Christidis, L., Ericson, P.G.P., Ohlson, J.I., Alström, P., 2020b.
An updated chronology of passerine birds. In: Fjeldså, J., Christidis, L., Ericson, P.G.P.
(Eds.), The Largest Avian Radiation: the Evolution of Perching Birds, or the Order
Passeriformes. Lynx Edicions, Barcelona, pp. 387–396.
Swainson, W., 1827. On several groups and forms in ornithology, not hitherto defined. Zoological Journal 3, 343–363.
Taylor, S.A., Larson, E.L., 2019. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3, 170-177.
Thompson, J.D., Higgins, D.G., Gibson, T.J., 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specifc gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673-4680.
Vigors, N.A., 1825. Sketches in ornithology; or, observations on the leading affinities of some of the more extensive groups of birds. Zoological Journal 2, 368–405.
Zhang, C., Rabiee, M., Sayyari, E., Mirarab, S., 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19, 153.
Zhang, D., Tang, L., Cheng, Y., Yao, H., Xiong, Y., Song, G., et al., 2019. ‘Ghost introgression’ as a cause of deep mitochondrial divergence in a bird species complex. Mol. Biol. Evol. 36, 2375-2386. .
Zhang, D., Rheindt, F.E., She, H., Cheng, Y., Song, G., Jia, C., et al., 2021. Most genomic loci misrepresent the phylogeny of an avian radiation because of ancient gene flow. Syst. Biol. 70, 961-975. .
Table
1.
Proposed species-level taxonomy for the family Alaudidae, comprising all lark taxa included in the present study in linear sequence (following the principles of Fjeldså et al., 2020b), compared to the taxonomy according to Gill et al. (2022) and Clements et al. (2022). In cases where the latter two are conflicting, the treatment supported by our study is highlighted in green font and the unsupported one in red font. Changes in our proposed taxonomy are highlighted in blue font; tentative but uncertain species status in purple font. The family, as defined here, contains 103 species across three subfamilies.
Table
2.
Divergence time estimates in million years based on the multilocus data (cf. Fig. 2) or, when available, based on the SNP data. Names in red font are treated as separate species by Gill et al. (2022), see Table 1.
Table
3.
Divergence time estimates in million years for the Mirafra africana–M. sharpii–M. hypermetra–M. somalica–M. ashi complex (A) and Eremophila complex (B), based on the multilocus data (cf. Fig. 2) or, when available, based on the SNP data (after a slash symbol; cf. Fig. 3); 95% highest posterior distributions are given in parentheses. Taxa that are believed to be marginally or more widely sympatric are highlighted with blue text (except for M. ashi–M. somalica rochei, which are sympatric, indicated by #; § indicates altitudinal, and hence also habitat, segregation), and taxa that are proposed to be treated as subspecies by Alström et al., (submitted) are highlighted with brown text.


 DownLoad:
DownLoad:







 Email Alerts
Email Alerts RSS Feeds
RSS Feeds